Expanded Access to Investigational Drugs
The Food and Drug Administration's Expanded Access Program allows for drugs that are investigational (i.e., not FDA approved) to be used for treatment use outside of a clinical trial. Sometimes this is referred to as compassionate use. Treatment use is not considered a clinical investigation, however, FDA submission and IRB review are necessary. The criteria for Expanded Access are largely determined by (1) the seriousness of a patient's condition, (2) other available FDA-approved treatment options, (3) patient population size needing treatment, and (4) drug status.
This webpage is a summarized guide to the FDA regulations on Expanded Access to Investigational Drugs for Treatment Use, found in 21 CFR 312.300. It does not provide full guidance for Expanded Access to Investigational Drugs; as such, FDA regulatory materials must also be consulted for full information.
Evaluating and Securing Expanded Access to an Investigational Drug
Step 1: Evaluate the seriousness of the patient's or patients' condition using the FDA's criteria
Step 3: Determine if the Expanded Access requires a new IND or can be amended to an existing IND
Step 4: Submit an Expanded Access submission to the FDA
Step 5: Submit an application for IRB review
Step 1: Evaluate the seriousness of the patient's or patients' condition using the FDA's criteria
All of the following criteria must be met, as described in 21 CFR 312.305(a):
-
- The patient or patients to be treated have a serious or immediately life-threatening disease or condition, and there is no comparable or satisfactory alternative therapy to diagnose, monitor, or treat the disease or condition;
- The potential patient benefit justifies the potential risks of the treatment use and those potential risks are not unreasonable in the context of the disease or condition to be treated; and
- Providing the investigational drug for the requested use will not interfere with the initiation, conduct, or completion of clinical investigations that could support marketing approval of the expanded access use or otherwise compromise the potential development of the expanded access use.
Step 2: Determine the patient population size that requires treatment use and evaluate the drug status using the FDA's criteria
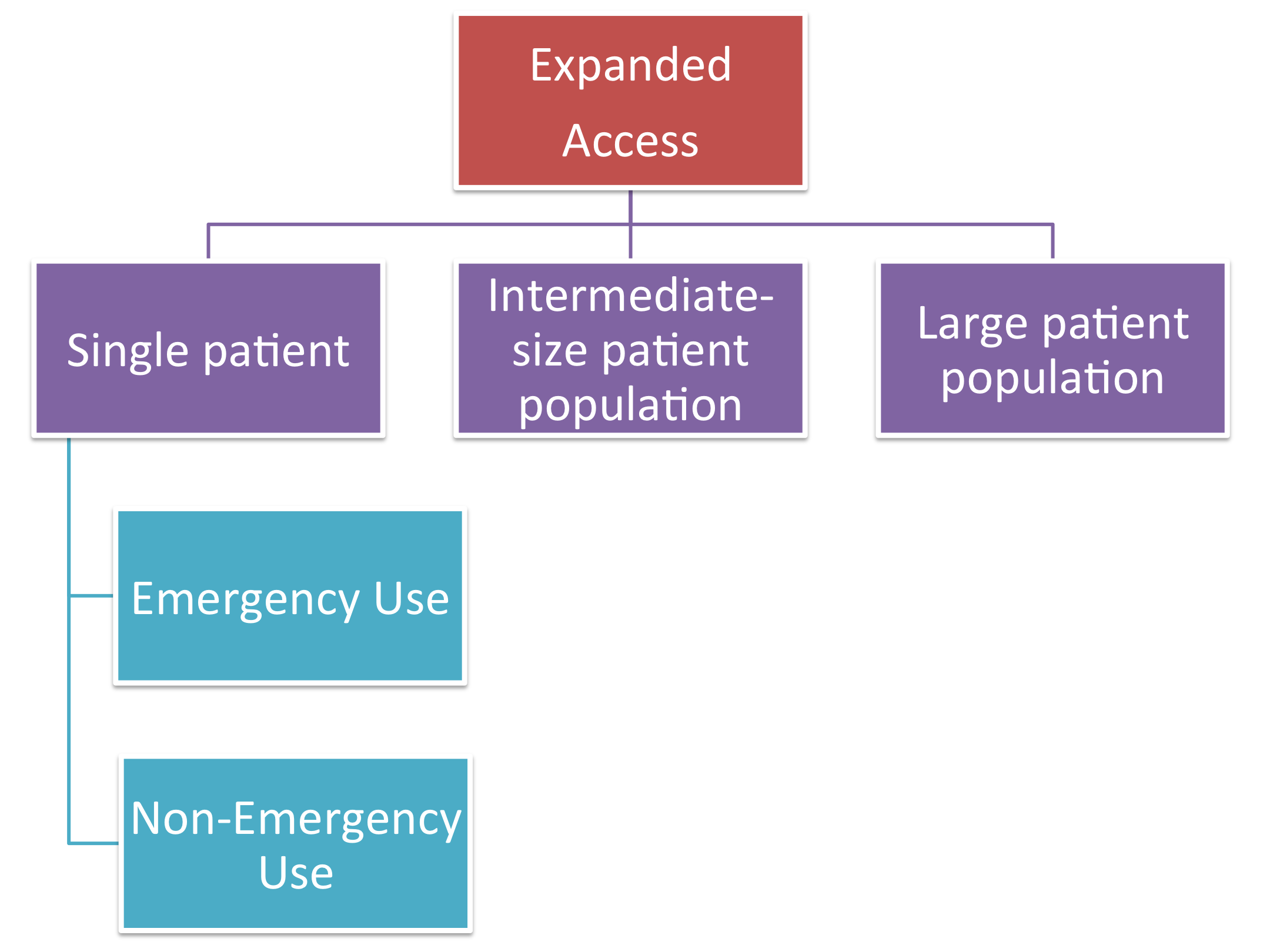
Single Patient Emergency Use
-
- Definition: 21CFR 56.102 (d); the patient is in an immediately life-threatening situation
- Criteria: 21 CFR 312.310 (a)
- Drug status: The patient cannot obtain the drug under another IND or protocol (21 CFR 312.310 (a)).
- University of Utah Guidance
Single Patient Non-Emergency Use
-
- Criteria: 21 CFR 312.310 (a); the patient has a serious condition and is not in an immediately life-threatening situation
- Drug status: The patient cannot obtain the drug under another IND or protocol (21 CFR 312.310 (a)).
Intermediate-Size Patient Population
-
- Definition: 21 CFR 312.315; the investigational use of a drug for the treatment of a patient population smaller than that typical of a treatment IND or treatment protocol
- Criteria: 21 CFR 312.315(b)
- Drug status: 21 CFR 312.315(a)
Large Patient Population, i.e. Treatment IND or Treatment Protocol
-
- Criteria and drug status: 21 CFR 312.320(a)
Step 3: Determine if the Expanded Access requires a new IND or can be amended to an existing IND
An IND is required for all Expanded Access. An individual physician may apply to the FDA for an IND or may function under an existing IND held by another physician or the drug manufacturer. Some IND holders will not allow for Expanded Access to be amended to their existing IND; in this case, the physician must apply for a new IND. Consult with the IND holder to determine if an existing IND can be amended.
Step 4: Submit an Expanded Access submission to the FDA
Use the instructions provided in 21 CFR 312.305(b) for the Expanded Access submission. These instructions must be followed for both new IND submissions and amendment submissions to existing INDs.
Step 5: Submit an application for IRB Review
For single patient emergency use, follow the Emergency Use of a Test Article instructions.
For all other expanded access options, submit a full new study application to the IRB. The application must include the following:
-
- IND documentation from the FDA/drug manufacturer
- Drug information via an Investigator's Brochure or a package insert
- An informed consent document
In non-emergencg situations, treatment may not begin until the IRB has approved the Expanded Access protocol.
Step 6: After initiating the treatment, ensure that all case histories and adverse events are properly recorded and reported
21 CFR 312.300 describes the safeguards and records that must be in place to ensure appropriate monitoring of patient safety. The requirements are different for the type of Expanded Access approved, based on patient population size. Ensure you are familiar with these requirements and can maintain appropriate records and oversight for Expanded Access protocols.